����(d��ng)ǰλ�ã������(zh��n)�е��������t(y��)Ժ >> ˎ���R��ԇ�(y��n)�C(j��)��(g��u) >> ���(xi��ng)���� >> �g�[����
�о��߰l(f��)���R���о����\(y��n)�й����(bi��o)��(zh��n)����Ҏ(gu��)��
�r(sh��)�g��2024��01��24����Ϣ��(l��i)Դ����վԭ��(chu��ng)
�����о��߰l(f��)����R���о���Investigator Initialed Trial,IIT����ָ�о����\(y��n)��������ˎƷ���t(y��)����е���\��ԇ�����_չ���R���о���ԓ��о������w/ ���ߞ��^�쌦(du��)�����漰�����A(y��)�����\�����ί����A(y��)����(f��)��̽����ͨ���ɱ�Ժ�ڍ��t(y��)��(w��)�ˆT�l(f��)��Ҳ�����c��I(y��)���f(xi��)��(hu��)��Ժ���λ(li��n)�ϰl(f��)��
�������ա�ˎ���R��ԇ�(y��n)�|(zh��)������Ҏ(gu��)������ICH-GCP Ҫ������(gu��)��(n��i)�����R���о���Ҏ(gu��)����Ҫ�����Y(ji��)����Ժ�ľ��w��r��Ҏ(gu��)�� IIT �Č�(sh��)ʩ���R���о�����(du��) IIT ��(sh��)�з���ּ�(j��)�������R���о����Ո(q��ng)���ո��һ������Ҫ�о��ߣ�PI�����ա�PI ����ָ�����_չ�R���о�������
����1.���(xi��ng)��(zh��n)��
����1.1 �о��������O(sh��)Ӌ(j��)��
������Ҫ�о��ߣ�Principal Investigator, PI��ؓ(f��)؟(z��)�о��������о���Ҫ�ļ����ƶ�����(y��ng)��Ո(q��ng)�y(t��ng)Ӌ(j��)/���в����҅��c���� 5010 �(xi��ng)Ŀ/308 Ӌ(j��)���ȣ�����ԃ���о����������Ʌ��Շ�(gu��)��(n��i)��J(r��n)���O(sh��)Ӌ(j��)Ҏ(gu��)����Ժ��“�R���о������M��ָ��”��
����1.2 �о��F(tu��n)�(du��)�ĽM����
����PI����(j��)�(xi��ng)Ŀ�ľ��w��r�M���о�С�M���о��F(tu��n)�(du��)���t(y��)�o(h��)�ˆT�����ܽ��|��ԇ�ߵ����P(gu��n)�ˆT����������P(gu��n)GCP��Ӗ(x��n)�����@���C����
����1.3 �о��ߕ�(hu��)�h�����_��
�����������İl(f��)��Ķ������R���о����h���_�о��ߕ�(hu��)�h���ռ����������Č�(du��)�����͌�(sh��)ʩ�����ԵĽ��h����Ҋ(ji��n)���R���о����k����ҕ��r���˅���(hu��)��
����1.4 ���(xi��ng)�Y�ϵ��ύ��
����1.4.1 �о����P(gu��n)�ļ��������PI/CRC ���գ�����1�� ��(zh��n)���Y�����ύ���C(j��)��(g��u)�k�����У�����2������3��PI��(ji��n)�v�躞���Ͻ�ԭ������
����1.4.2 �Y�ϵ���ʽ���顣GCP�k�����ؕ��M(j��n)����ʽ����������ϸ��o��“�C(j��)��(g��u)����̖(h��o)”��
����1.4.3 ���|(zh��)�Y�ϵ��Ͻ�������ʽ����ͨ�^(gu��)���ڸ���4�����“�C(j��)��(g��u)����̖(h��o) ”���f�����|(zh��)�����1���������k���ң����ϕ�(hu��)ӑՓ��
����1.4.4 ����ע�����(xi��ng)��
����1.4.4.1 �о������ȸ���(j��)[���һ]���о����“��� 2 ”���Y����r��֪�R(sh��)�a(ch��n)��(qu��n)����ԇ���r��/�a(b��)��?sh��)����о��������о���ͬ���������_��
����1.4.4.2 �煢��������λ�Ķ������R���о����о��ߑ�(y��ng)�ύ�M�L(zh��ng)��λ�Ă��������ͽ�(j��ng)��������(zh��n)���о�������֪��ͬ�����
����1.4.4.3 ������Ո(q��ng)�f(xi��)��(hu��)/�W(xu��)��(hu��)�Y�����R���о��(xi��ng)Ŀ����Ҫ�t(y��)Ժ/���ĺ��µ�����(y��ng)���R���о����k�����k�����P(gu��n)���m(x��)�����(xi��ng)Ŀ�@���Y����(y��ng)�������M(j��n)����ʽ���(xi��ng)��
����1.4.4.4 ��(du��)�L(f��ng)�U(xi��n)��(j��ng)�M(f��i)��Ŀ�ԺУ/�����R���о�����(y��ng)�õ�PI���ڿ������κ���ͬ����
����1.4.4.5 ��(du��)����Ϣ�{(di��o)����о����漰��ˎ/�\����Ϣ���t(y��)���M(f��i)�õ�������Ϣ�ģ������t(y��)��(w��)̎������
����1.4.4.6 �漰�����t(y��)���O(sh��)���IIT�(xi��ng)Ŀ����(y��ng)���ȵõ�PI���ڿ������κ���ͬ����ͬ�r(sh��)Ҫ�ύ����(w��)̎���t(y��)��(w��)̎����������ԡ�
����2.���(xi��ng)����
����GCP�k���Ұ��ա����(xi��ng)���˵� SOP���M(j��n)����K�Y(ji��)�����顣
����3.���팏��
����3.1 �о��߰��Ղ���ί�T��(hu��)��Ҫ���(zh��n)����� �����“�R���о�����ƽ�_(t��i)”�����(b��o)�����M(j��n)�Ђ����f����ͬ�r(sh��)�f�����|(zh��)����ϵ������k���ҡ�
����3.2 ��K��“����ί�T��(hu��)������”���о��߱�����ԓ�(xi��ng)Ŀ���о��ļ��A����
����4.��ͬ����
����4.1 �R���о���ͬ�����(xi��ng)��PI/CRC ���ա��R��ԇ�(y��n)��ͬ��ӆ�� SOP���c�Y�����ͷ����ĔMӆ��ͬ�l��f(xi��)�̽�(j��ng)�M(f��i)�A(y��)������“�R���о�����ƽ�_(t��i)”�ύ��������ĺ�ͬ/�f(xi��)�h��
����4.2 ��ͬ����ͨ�^(gu��)������GCP�k�����ؕ���Ժ�L(zh��ng)���ڙ�(qu��n)�˺�����Ч��
����4.3 ��ͬ��ʽ������������_չ�R���о���
����4.4 ���(xi��ng)Ŀ���(xi��ng)�r(sh��)�o(w��)�κν�(j��ng)�M(f��i)�Y����PI�躞��“�o(w��)�κν�(j��ng)�M(f��i)�Y������ ”�� ����5�������ڌ�(sh��)ʩ�^(gu��)�������@�ý�(j��ng)�M(f��i)�Y����PI��(y��ng)�����R��ԇ�(y��n)��ͬ��ӆ�� SOP�� �k�����P(gu��n)������
����5.“���z�k ”������Ո(q��ng)���(xi��ng)����������Ո(q��ng)“���z�k ”�������(xi��ng)Ŀ��PI/CRC ��(y��ng)��C(j��)��(g��u)�k���������Ո(q��ng)�����ա����z�k��Ո(q��ng)����ָ�����k����
����6.�(xi��ng)Ŀ��(sh��)ʩ
����6.1 �о��Y�ϵĜ�(zh��n)��
��������(d��ng)��(hu��)���_ǰ��PI/CRC ��(y��ng)�_���R���о��Y�Ϝ�(zh��n)����(d��ng)��
����6.2 �о�ˎƷ����е�ȵĜ�(zh��n)��
��������(d��ng)��(hu��)���_ǰ���о�ˎƷ����е���ṩ�ߌ����P(gu��n)���Y�����о�С�M��PI/CRC���ա�ˎ������ƶȡ���ˎ��Ľ��ա��������ְl(f��)����������߀���N���� SOP�����Ɍ���ؓ(f��)؟(z��)���Y�Ľ������������ְl(f��)�����պ���߀��
����6.3 ����(d��ng)��(hu��)�����_��
����PI���ա�ˎ���R��ԇ�(y��n)�(xi��ng)Ŀ����(d��ng)�� SOP���������_�(xi��ng)Ŀ����(d��ng)��(hu��)��
����6.4 ����؟(z��)�εĽ綨��
�����(xi��ng)Ŀ���팍(sh��)ʩPIؓ(f��)؟(z��)����PI��(du��)��ԇ�ߙ�(qu��n)�����t(y��)����ȫؓ(f��)ȫ؟(z��)����(du��)�о���(sh��)��(j��)���挍(sh��)��ؓ(f��)ȫ؟(z��)��ֱ��؟(z��)�������ڿ���ؓ(f��)؟(z��)�˼��R���о���ؓ(f��)�й���؟(z��)���������g��؟(z��)������
����6.5 �ֹ����ڙ�(qu��n)��
�������c���о��ˆT������ʹ؟(z��)��(y��ng)���ϸ��Ԉ�(zh��)�I(y��)�������ڙ�(qu��n)��(n��i)�����漰֪��ͬ�����t(y��)���Д����t(y��)�ڵȭh(hu��n)��(ji��)����ɱ�Ժע��(c��)����(j��ng)PI�ڙ�(qu��n)���R���t(y��)��ؓ(f��)؟(z��)��(zh��)�����R���о����P(gu��n)�t(y��)�����v���ĕ��ĕ���������PI�ڙ�(qu��n)���R���t(y��)�������_�J(r��n)���о��ˆT�����Ї�(gu��)���P(gu��n)GCP��ICH-GCP���о����������P(gu��n)SOP��(sh��)ʩ�R���о����硶��ԇ��֪��ͬ���SOP����ԭʼ�Y��ӛ䛵� SOP����������(b��o)������SOP���������¼�����(y��n)�ز����¼�̎���cӛ䛵�SOP����ˎ���R��ԇ�(y��n)SAE��(b��o)��� SOP���ȣ���
����6.6 SAE ���ψ�(b��o)��
�������о��^(gu��)���������l(f��)�������¼����о��߰��շ������t(y��)Ժ���P(gu��n)Ҏ(gu��)���e�O̎��������(y��n)�ز����¼���(y��ng)���r(sh��)��(b��o)��?zh��n)���ί�T��(hu��)��������ˎƷ����ˎƷ��������(y��ng)��(b��o)��ͱO(ji��n)�y(c��)�����k������Ҫ������(b��o)�汾Ժ��������(y��ng)�O(ji��n)�y(c��)���ģ�ˎ�����R��ˎ�W(xu��)�ң����������t(y��)����е����ˎƷ��������(y��ng)��(b��o)��ͱO(ji��n)�y(c��)�����k�������t(y��)����е�O(ji��n)������?xi��ng)l������Ҫ��(b��o)�汾Ժ�t(y��)��(w��)̎���P(gu��n)ؓ(f��)؟(z��)����
����7.�|(zh��)������
����7.1 �о��ߑ�(y��ng)�ƶ�����(y��ng)���(xi��ng)ĿSOP�Ա����о��(xi��ng)Ŀ��Ҏ(gu��)����(sh��)ʩ��
����7.2 �������Ğ�M�L(zh��ng)��λ��PI��(y��ng)��(du��)�����ĵ��о��|(zh��)���M(j��n)�б�Ҫ�ıO(ji��n)����
����7.3 ���о��(xi��ng)Ŀ�ж���c�����Y��������(y��ng)���պ�ͬ�ļs��̎��������؟(z��)��(qu��n)����ԇ�ߓp�����r��/�a(b��)����
����7.4 �R���о����|(zh��)������Tҕ���w��r��(du��)�о��(xi��ng)Ŀ�|(zh��)�����M(j��n)���M(j��n)�Йz������(du��)���ڵĆ�(w��n)�}�l(f��)��“��(w��n)�}ͨ���”���о����������IJ�����؏�(f��)�����ڇ�(y��n)�؆�(w��n)�}������Ҫ�r(sh��)�l(f��)��“������”����(du��)�`�� GCP �ͷ�������ɇ�(y��n)�غ���������ա��R���о�ȱ�ݹ����ƶȡ������P(gu��n)���T�M(j��n)��̎����
����7.5 �(xi��ng)Ŀ��(zh��)���^(gu��)���У��о��ߑ�(y��ng)�e�O��ϸ�����z������(du��)���ڵĆ�(w��n)�}���r(sh��)���ġ�
����8.�Y�ϱ���/���Y(ji��)/�l(f��)��
����8.1 �(xi��ng)Ŀ�Y(ji��)���������ՙC(j��)��(g��u)���n�������ƶȡ����� PI/CRC ؓ(f��)؟(z��)�о��Y�ϵ����������б��档
����8.2 �R���о����Y(ji��)��(b��o)������GCP�k���Һ�����PI��(y��ng)��(du��)���Y(ji��)��(b��o)����挍(sh��)�ԺͿƌW(xu��)��ؓ(f��)؟(z��)���_�J(r��n)��������GCP�k�����ؕ�����GCP���Ό�醺��w“GCP�k����”�¡�
����8.3 PI �� �l(f��) �� Փ �� ֮ ǰ ��(y��ng) �� �� �� ��(sh��) ��(j��) �� RDD ƽ �_(t��i) �� �� �������w�Ʌ��ա��P(gu��n)���о�Փ����Ҫ�䰸�о���(sh��)��(j��)��֪ͨ���[�����Σ�2017��45 ̖(h��o)������
�������һ������R���о��ķ��������
������һ�ע��(c��)��R��ԇ�(y��n)
����1. ����(j��)ˎ�O(ji��n)�������T�Ĺ������
����1.1 ˎ���R��ԇ�(y��n)���֞� I �ڡ�II ����III �ں� IV ��
����1.2 �t(y��)����е�R��ԇ�(y��n)
����1.3 �w���\��ԇ���R��ԇ�(y��n)
�����ڶ���о��߰l(f��)����R���о�
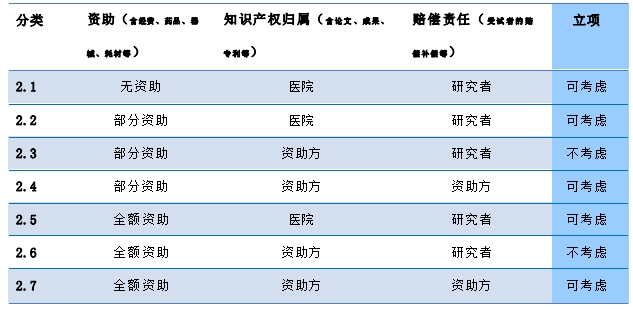
����2. �����Y����r��֪�R(sh��)�a(ch��n)��(qu��n)�w���Լ���ԇ�ߓp���r��؟(z��)�η��
�����Y����(l��i)Դ������ˎ���������(hu��)���W(xu��)��(hu��)/�f(xi��)��(hu��)�������A�t(y��)�W(xu��)��(hu��)���Ї�(gu��)�����f(xi��)��(hu��)�����Aƽ����ȣ���˽�˾�ٛ(z��ng)�������Y����(l��i)Դ�鱾���������t(y��)Ժ�������п��]��
����3. �����о��O(sh��)Ӌ(j��)��ͷ��
����3.1 ���A(y��)�����Interventional study��
����3.2 �\�������Diagnostic study��
����3.3 �^�������Observational study��
����4. �����m��(y��ng)�Y�������
����4.1 �m��(y��ng)�Y������(n��i)���о���ˎƷ��(y��n)���f(shu��)����ʹ�������漰��׃�f(shu��)�����m��(y��ng)�Y�������oˎ;�����oˎ��ʽ���r(sh��)�g����
����4.2 �����m��(y��ng)�Y�о����[�����I(y��)�_չ����о���(y��ng)���[���I(l��ng)�����m��(y��ng)�Y��̽�����о����磺���������N�������½oˎ�������������ڻ�������ˎ��������/��/ һ����ˎ�����ӆ�ˎ��(li��n)����ˎ�����Ӄ�ͯ��ˎ�������ˎ�����Ʌ�����CFDA 2012���C���ġ������п��[��ˎ���������m��(y��ng)�Y���g(sh��)ָ��(d��o)ԭ�t�����ѽ�(j��ng)���еĿ��[��ˎ�����ӷ��[���I(l��ng)�����m��(y��ng)�Y����Ո(q��ng)������ע��(c��)��о���
����5. ������L(f��ng)�U(xi��n)���
����5.1 ���L(f��ng)�U(xi��n)�о������A(y��)��ʩ�鳬�m��(y��ng)�Y������ˎ�������g(sh��)ʽ��(chu��ng)���ԙz�����
������ͯ����������g���ߣ���65 �q�����о���(du��)����о���
����5.2 �е��L(f��ng)�U(xi��n)�о������ڸ��L(f��ng)�U(xi��n)�c���L(f��ng)�U(xi��n)���о�
����5.3 ���L(f��ng)�U(xi��n)����^�������











���l(w��i)��.jpg)
